Home / Articles / What tests should I take to check the functioning of the adrenal glands?
The adrenal glands are small paired organs of the endocrine system that regulate many processes in our body. The state of immunity, weight gain and the tendency to develop fatty deposits in certain places, emotional stability, stress resistance, urination and metabolism depend on their work. The paired organs are located deep in the tissues, so they are almost invisible on ultrasound. An effective way to assess their condition is laboratory diagnostics. Find out what tests you need to take to check your adrenal glands and when you need to undergo such testing.
When should you take an ACTH test?
- Sudden and pronounced change in weight.
- Migraine, back pain.
- Fatigue, lethargy, apathy, depression, insomnia.
- Acne in adults.
- Increased body hair in women, disruption of the menstrual cycle.
- Hyperpigmentation of the skin in areas not exposed to ultraviolet radiation.
- Early onset of puberty.
- Deviation from the norm in the concentration of cortisol in the blood.
- Treatment with glucocorticosteroids for a long period.
- Suspicion of pituitary adenoma.
- Diabetes.
- Osteoporosis, discomfort, bone pain, crumbling teeth.
- Infertility.
1) Dehydroepiandrosterone sulfate
Dehydroepiandrosterone sulfate (DHEA-s, DEA-SO4) is a steroidal androgenic hormone synthesized by the adrenal cortex. It is used by the body to produce testosterone and estrogen. Its excess content can lead to the threat of termination of pregnancy and spontaneous miscarriage.
In what cases is a blood test for DHEA-s prescribed?
- adrenogenital syndrome;
- tumors of the adrenal cortex;
- ectopic ACTH-producing tumors;
- habitual miscarriage;
- fetal hypotrophy;
- delayed puberty;
- diagnosis of the state of the feto-placental complex from 12-15 weeks of pregnancy.
How to prepare for a blood test for DHEA-s?
Before donating blood, it is recommended to avoid stress and fatigue, limit physical activity, and refrain from frequent smoking. It is advisable to donate blood in the morning, on an empty stomach. If you need to donate blood at another time, you must abstain from eating for 4-6 hours.
What medications should not be taken before taking a blood test for DHEA-s?
Dexamethasone, hydrocortisone, prednisolone, diprospan, estrogens, oral contraceptives. When donating blood, you must inform the nurse about taking medications that affect the level of hormones in the blood.
What are the deadlines for completing the analysis?
1-2 business days.
What are the normal blood test for DHEA-s?
These values may vary depending on age, test systems and analyzers used:
- women: 810 – 8991 nmol/l;
- men: 3591 – 11907 nmol/l.
Detailed description of the study
ACTH is a pituitary hormone, the most important regulator of the function of the adrenal cortex.
Adrenocorticotropic hormone (ACTH) is a chemically active protein. It is produced by the anterior lobe of the pituitary gland.
With the bloodstream, the hormone reaches the target organ - the adrenal glands. By acting on their cortical layer (superficial part), adrenocorticotropin stimulates the release of adrenal hormones - cortisol, cortisone, corticosterone. Corticotropin indirectly affects the release of adrenaline, female and male sex hormones.
Biochemical and physiological effects of adrenocorticotropin:
1. Resisting stress. In situations of overstrain (emotional and physical overload, cold) it increases the concentration of the steroid hormone cortisol, which helps mobilize the body's resources.
2. Effect on brain structures. The psychoemotional state of a person depends on the increase or decrease in ACTH concentration. Lack of the hormone leads to depression, memory impairment, and suppression of cognitive functions.
3. Stimulation of the production of sex hormones. Under the influence of ACHT, the adrenal glands increase in volume and produce more progesterone, estrogen, and testosterone.
4. Participation in the process of cholesterol synthesis, which is necessary for the construction of cell walls, the formation of vitamin D, and red blood cells.
5. Increased skin pigmentation. Adrenocorticotropic hormone stimulates melanocytes, which produce the pigment melanin.
6. Regulation of weight, muscle volume. Adrenocorticotropin affects the increase or decrease in appetite, the removal of fat from subcutaneous tissue, and is involved in the regulation of glucose metabolism (accumulation of glycogen in muscles). Stimulates the release of insulin from pancreatic beta cells (along with other regulatory factors). Activates the absorption of amino acids and glucose into muscle tissue. The main causes of disruption of corticotropic hormone production:
- diseases of the pituitary gland: decreased (hypofunction) or increased (hyperfunction) activity;
- disruption of the adrenal cortex;
- abnormal external secretion of corticotropin due to the presence of a tumor in the body (ectopic corticotropin production syndrome, EPS).
Growth factors
IPFR I is one of the main factors important for the regulation of cell and tissue development processes. First of all, the concentration of the substance in the blood depends on age. The amount of somatomedin in the body of a child or adult is influenced by nutrition.
Tests for the determination of insulin-like growth factor make it possible to assess the somatotropic function of the pituitary gland. It is recommended to take a blood test as quickly as possible when assessing changes in metabolic status is needed, as well as in cases of growth retardation. IFRs help monitor the patient’s condition during the treatment of dwarfism and acromegaly.
Reduced levels of insulin-like growth factor are characteristic of the following conditions:
- dwarfism,
- hypopituitarism,
- hypothyroidism,
- anorexia,
- emotional deprivation syndrome,
- Laron dwarfism,
- liver failure,
- inflammatory bowel diseases.
An increased level of IGF is observed if there are the following diseases:
- acromegaly,
- Cushing's syndrome,
- renal failure.
Taking some medications also leads to an increase in the concentration of the substance in the body.
In the laboratory of the CDC “Health Laboratory” you can undergo a study to determine the amount of this compound, as well as somatotropic hormone (SH), necessary for the formation of bones, muscles and organs. It is recommended to conduct a growth hormone study when:
- slow or accelerated physical development,
- osteoporosis,
- muscle weakness,
- increased sweating,
- porphyria and other conditions.
Blood for examination is donated on an empty stomach. Recommendations for preparing for the study can be found on the website of the CDC Laboratory of Health.
References
- Nazarenko G.I. Clinical assessment of laboratory research results / G.I. Nazarenko, A.A. Kishkun - M.: Medicine, 2006 - 543 p.
- Allen MJ; Sharma S. Physiology, Adrenocorticotropic Hormone (ACTH) March 3, 2021 URL: https://www.ncbi.nlm.nih.gov/books/NBK500031/ (accessed 01/10/2020)
- Fish S., MD Adrenocorticotropic Hormone (ACTH), October 2019 URL: https://www.hormone.org/your-health-and-hormones/glands-and-hormones-a-to-z/hormones/adrenocorticotr… ( access date 01/10/2020)
Reasons for the development of pathology
The exact causes of adrenal adenoma are unknown to medicine. There are factors that provoke the development of pathology. These include:
- heredity (rare);
- smoking, alcoholism;
- obesity;
- frequent stressful situations;
- age over 50 years;
- exposure to physical and chemical carcinogenic factors;
- parasitism in the body of certain types of oncogenic viruses (papillomavirus, Epstein-Barr virus);
- hyperplasia of the adrenal cortex;
- endocrine tumors of other glands (parathyroid, pancreas, pituitary gland).
Procedure for collecting urine for research
To properly prepare the material, you need to perform the following steps step by step:
- Two days before the test, take a special preservative from the laboratory.
- Buy a sterile container for urine at the pharmacy.
- Wash thoroughly and scald with boiling water a large glass jar with a volume of at least 2.5 liters, closed with a tight lid.
- Place the preservative obtained in the laboratory into the jar.
- The next morning, release the first portion of urine into the toilet.
- Starting from the next desire to visit the toilet, during the day all biological fluid must be collected in a prepared jar, closing it each time.
- The container with the material should be kept in a cool, dark place.
- Mix all the material and pour approximately 50 ml into a container.
- Deliver the test to the laboratory no later than 1.5 hours after collection.
Catecholamine analysis
Sometimes, in cases of persistent hypertension, suspected myocardial infarction, or tumor processes in the adrenal glands, the doctor recommends determining the level of a special group of biological active substances - catecholamines.
To check the production of this group of hormones by the adrenal glands, it is necessary to take a catecholamine test. Their content is determined in venous blood and daily urine.
Blood analysis
When preparing for the study, you must follow the following rules:
- two days before the start of material collection, all diuretics are canceled, and medications are canceled one day before;
- the day before the analysis, you must give up smoking, nervous and physical stress.
Attention is also paid to the daily diet. For two days before the test, you should not drink tea, coffee, alcoholic beverages, or eat bananas, cheeses, or avocados. The last meal should be 12 hours before visiting the laboratory.
Analysis of urine
Urine can be collected for 3, 6, 12 hours and a full day. The longer the biological fluid is collected, the more reliable the research results will be.
Normal catecholamine levels:
| Hormone | Amount in urine | Amount in blood |
| Adrenalin | up to 21 mcg/day | up to 110 pg/ml |
| Norepinephrine | 15–80 mcg/day | from 70 to 750 pg/ml |
| Dopamine | 60–400 mcg/day | up to 87 pg/ml |
Serotonindo 200 mcg/day 50 to 220 pg/ml
Also, to control the production of hormones by the adrenal glands, in addition to the listed names of tests, the content of a number of electrolytes is determined.
Determining adrenal hormones will help not only in diagnosing the disease, but also in choosing the most effective treatment. To early detect pathology in the body and prevent the development of severe forms of disease, such tests should be carried out every few years.
Author: Irina Ramazanova, doctor, especially for Nefrologiya.pro
Relevance
Primary hyperaldosteronism (PHA) is the most common form of secondary arterial hypertension, detected in 4.3% of all patients suffering from high blood pressure and in 9.5% of patients referred for this reason to specialized centers [1]. The most common forms of PHA (up to 90% of all cases) are bilateral adrenal hyperplasia (idiopathic hyperaldosteronism, IHA) and unilateral aldosterone-producing adenoma (APA). Much less commonly, PHA is caused by unilateral adrenal hyperplasia, aldosterone-producing carcinoma, aldosterone-producing tumor of extra-adrenal location, as well as hereditary forms of familial hyperaldosteronism [2].
It is known that arterial hypertension is a predictor of cardiovascular morbidity and mortality [3]. Patients with PHA have an even greater cardiovascular risk than patients with essential arterial hypertension with the same degree of blood pressure increase, regardless of gender and age [4]. Among patients with PHA, there is an increased prevalence of insulin resistance, diabetes mellitus (DM), metabolic syndrome [5, 6], osteoporosis [7], depression and anxiety [8], conditions that are more likely a manifestation of excess gluco- rather than mineralocorticoids. This is of particular importance in light of recent studies showing that PHA is often associated with hypercortisolism syndrome. Since the first mention in the literature of the combined secretion of aldosterone and cortisol (1979), the number of reported cases of the so-called Connsching syndrome has increased. Analysis of recent data allows us to conclude that hypercortisolism in PHA is very common and is closely associated with an increased risk of metabolic disorders and cardiovascular complications [9]. Thus, in a study by G. Piaditis et al. [10] the prevalence of hypercortisolism associated with PHA among 83 incidentalomas was 12.1%. In a study by K. Hiraishi et al. [11] 8 out of 38 patients with PHA had subclinical Cushing's syndrome, and in a retrospective analysis of 414 cases of confirmed PHA, hyperproduction of cortisol was detected in 22 patients (5.31%) [12]. According to W. Arlt et al. [9], with combined secretion, the concentration of glucocorticoids in PHA was close to that in patients with subclinical hypercortisolism, who have an increased cardiovascular risk [13]. In both identified subtypes of PHA (unilateral APA and bilateral IHA), glucocorticoid secretion was significantly higher than in the control group, which included healthy individuals and patients with hormonally inactive adrenal formations. Aldosterone- and cortisol-secreting tumors can be either benign (aldosterone- and cortisol-producing adenoma (A/CPA)) or malignant (aldosterone- and cortisol-producing adenocarcinoma), differing in size, cellular composition and hormonal activity [14]. Thus, the traditional distinction between Cushing's and Conn's syndromes is not as clear as previously thought.
Methodology for searching primary sources
A search of literature reviews and case reports published up to 2021 on combined aldosterone and cortisol hypersecretion and the role of hybrid steroids (18-oxocortisol and 18-hydroxycortisol) in the diagnosis of adrenal neoplasms was carried out through the MEDLINE database using PubMed. The search was carried out using the PubMed Search Builder tool, which included the following combinations of MeSH terms: “Hyperaldosteronism and Cushing syndrome”; "Cushing Syndrome and Aldosterone"; "Hyperaldosteronism and Hydrocortisone"; “Aldosterone and Hydrocortisone and Adenoma”, “Aldosterone and Hydrocortisone and Adrenal Gland Neoplasms”; "18-oxocortisol"; "18-hydroxycortisol." When relevant papers were found, an additional search was carried out through the “References”, “Similar articles” and “Cited by other articles” sections.
Characteristic features: greater risks, diagnostic difficulties
The increased risk of cardiovascular complications in PHA is undeniable. An increased incidence of stroke in patients with PHA has been identified in both sporadic and familial forms [15] and is partly explained by the direct effects of aldosterone, inducing inflammation, fibrosis and vascular remodeling [16, 17]. In in vivo
It has been shown that activation of mineralocorticoid receptors (MCRs) leads to remodeling of cerebral vessels and aggravates the consequences of cerebral ischemia [18]. Animal models have demonstrated the involvement of aldosterone and MCR in the provocation of ventricular arrhythmias and atrial fibrillation [19, 20]. The role of aldosterone in the pathogenesis of heart failure is confirmed not only by its direct effect on cardiomyocytes, but also by the protective effect of MCR antagonists in patients with decreased ventricular function [21].
The results of studies on the relationship between hyperaldosteronism and diabetes are controversial. However, G. Hanslik et al. [6], based on an analysis of the literature and their own research, come to the conclusion that there is an increased risk of impaired glucose tolerance, diabetes, and metabolic syndrome in patients with PHA. The Expert Committee on the Diagnosis and Classification of Diabetes Mellitus of the American Diabetes Association also classifies PHA as a risk factor and endocrine cause of diabetes [22]. Among the mechanisms of impaired glucose metabolism in PHA, the following are considered: increased inflammation, oxidative stress, decreased mass of β-cells of the islets of Langerhans and their functional activity, caused by the direct effect of aldosterone [23–25], its possible effect on the sensitivity of peripheral tissues to insulin, as well as diabetogenic effect of hypokalemia [26].
Patients with hypercortisolism are also characterized by hypertension, an increased risk of developing cardiovascular and metabolic disorders, including morbid obesity, impaired glucose tolerance, diabetes, spinal osteoporosis and hypercoagulability [27].
Aldosterone and cortisol have similar affinities for MCRs. Normally, the concentration of free cortisol in the blood significantly exceeds the concentration of aldosterone; therefore, a significant proportion of MCRs are bound by cortisol, but under physiological conditions MCRs are not activated due to special inhibitory intracellular systems such as 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2). However, cortisol is still able to activate the MCR [28, 29]. Thus, MCR blockade reduces the severity of changes in the cardiovascular system even in patients with low and normal levels of aldosterone and renin [30, 31]. At high concentrations and under certain conditions, cortisol can activate MCRs expressed in the cardiovascular system [32], and high concentrations of aldosterone and cortisol are independent risk factors for cardiovascular events in patients with chronic heart failure [30]. Adding to the negative effects of hyperaldosteronism, increased cortisol secretion may increase the risk of metabolic and cardiovascular complications. The summation of the negative effects of excess secretion of aldosterone and cortisol is confirmed by K. Hiraishi et al. [11], who found that patients with A/CPA have a greater risk of cardiovascular disorders, including atrial fibrillation, myocardial infarction, heart failure, coronary atherosclerosis and stroke, and diabetes, than patients with PGA without cortisol hypersecretion.
Another concern for patients with A/KPA is the increased risk of adrenal insufficiency after removal of an adrenal mass. In a review of studies published from 1980 to 2013, the prevalence of postoperative adrenal insufficiency among persons undergoing unilateral adrenalectomy for corticosteroma was 65.3% among 248 patients with subclinical hypercortisolism and 99.7% among 377 patients with full-blown Cushing's syndrome. Restoration of normal cortisol secretion was more often observed among individuals with a subclinical form - 138 out of 141 (97.9%) than in patients with overt Cushing's syndrome - 351 out of 376 (93.4%) [33]. In a review by M. Späth et al. [14] analyzed cases of APA with subclinical hypercortisolism after removal of the adrenal gland with a tumor. In two cases where autonomic cortisol hypersecretion was not diagnosed preoperatively, a hypoadrenal crisis developed; in six others, even when subclinical hypercortisolism was diagnosed before adrenalectomy and oral glucocorticoid replacement therapy was prescribed after its implementation, patients experienced severe symptoms of adrenal insufficiency. The data presented indicate that overproduction of cortisol in such patients is likely associated with an increased risk of adrenal insufficiency after surgical treatment. All of the above requires caution regarding the diagnosis of aldosterone- and cortisol-secreting tumors. More detailed research is needed.
The most important diagnostic problem in A/CPA turned out to be the risk of false interpretation of the results of comparative selective venous blood sampling from the adrenal veins (CVBD). According to the Russian and European clinical guidelines for PHA, differential diagnosis of PHA forms with unilateral and bilateral aldosterone overproduction is fundamental for choosing treatment tactics: in APA, the treatment of choice is unilateral adrenalectomy, while patients with IHA receive lifelong therapy with MCR antagonists. CVBD is recognized as the “gold standard” in the lateralization of aldosterone overproduction [34, 35]. However, the classic CVD protocol, which relies on venous cortisol concentrations to confirm the accuracy of catheterization, carries the risk of misinterpretation of the results in patients with subclinical hypercortisolism. Analysis of the results of CVBD in patients with aldosterone- and cortisol-producing adenoma revealed the risk of inadequate lateralization of aldosterone overproduction when calculating the aldosterone/cortisol ratio. Therefore, the CVD protocol for patients with or suspected hypercortisolism has been considered unreliable [36]. The possibility of using modified variants of CVID in patients with A/CPA is being considered, among which an assessment of selectivity based on the plasma concentration of aldosterone as such without correction for the value of cortisol concentration [37] and an assessment of selectivity based on the concentration of methylated catecholamines [38] were proposed, as well as to the search other means of differential diagnosis of unilateral and bilateral forms of PHA [39].
Possible causes of combined secretion
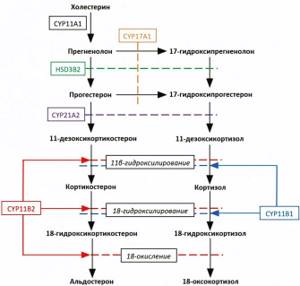
When considering the likely mechanisms of cortisol hyperproduction in PHA, one should proceed from the histological features of APA. It is known that the final stages of the biosynthesis of gluco- and mineralocorticoids in the human adrenal cortex are catalyzed by two mitochondrial cytochrome P450 enzymes that are 93% homologous in amino acid sequence: CYP11B1 (11β-hydroxylase) and CYP11B2 (aldosterone synthase), respectively [40]. In the zona fasciculata, 11-deoxycortisol, a steroid precursor hydroxylated at C17 in the early stages of steroidogenesis by the enzyme CYP17A1 (17α-hydroxylase), is again hydroxylated at position 11b by CYP11B1, turning into cortisol. In the zona glomerulosa, where the activity of 17α-hydroxylase is suppressed, aldosterone synthase sequentially catalyzes the reactions of 11β-hydroxylation, 18-hydroxylation and 18-oxidation of 11-deoxycorticosterone. The last step leads to the conversion of 18-hydroxycorticosterone (18OHB) to aldosterone (Fig. 1, see Fig.
Rice.
1. Pathways of steroidogenesis leading to the synthesis of aldosterone, cortisol, as well as hybrid steroids: 18-oxocortisol and 18-hydroxycortisol. CYP11A1 – 20,22-desmolase (“Cholesterol side chain degrading enzyme”), HSD3B2 – 3-beta-hydroxysteroid dehydrogenase type 2, CYP17A1 – 17α-hydroxylase, CYP21A2 – 21-hydroxylase, CYP11B1 – 11β-hydroxylase, CYP11B2 – aldosterone synthase . Section “Additional Information”) [41]. In unchanged adrenal glands, aldosterone production is possible only in the zona glomerulosa of the adrenal gland, where cells carrying the CYP11B2
, encoding aldosterone synthase, are localized, while the expression of the enzymes necessary for the synthesis of cortisol - 17α-hydroxylase and 11β-hydroxylase, encoded by the
CYP17A1
and
CYP11B1
, respectively, occurs in beam zone [42, 43]. Restriction of the expression of CYP17A1 and CYP11B1 to the zona fasciculata (in the case of CYP17A1, also to the reticularis), and CYP11B2 to the glomerular one, leads to a functional separation of the sites of cortisol and aldosterone synthesis [44].
APAs composed exclusively of zona glomerulosa cells are rare and typically consist of various cell types, predominantly similar to zona fasciculata cells, as well as less numerous “hybrid” cells having cytological characteristics of both zona glomerulosa and zona fasciculata [45, 46]. ]. The histological heterogeneity of APA tissue containing cells expressing CYP17A1 and CYP11B1 [12] underlies its potential for cortisol secretion, which is consistent with the results of in vitro
[47]. M. Späth et al. [14] put forward three hypotheses for the causes of hyperproduction of cortisol in APA. First, the tumor may have a larger proportion of cortisol-producing cells than “pure adenomas,” which consist almost entirely of zona glomerulosa cells. This theory could be confirmed by data on different levels of 11β-hydroxylase (CYP11B1) in APA tissue depending on the degree of glucocorticoid hypersecretion. However, the results of such studies are mixed. If L. Tang et al. [12] using quantitative real-time PCR did not reveal a significant difference in the expression of 11β-hydroxylase between A/KPA and “pure” aldosterones, then W. Arlt et al. [9] using quantitative immunohistochemical analysis found a clear relationship between intratumoral enzyme expression and daily excretion of glucocorticosteroids.
Secondly, the number of cortisol-producing cells, while maintaining their share in the total mass of adenoma tissue at the same level, can become clinically significant due to the rather large size of the formation [14]. It has been shown that the combination of hypercortisolism and PHA most often occurs in the formation of the adrenal gland, the size of which exceeds that of the average aldosterone. In a study of 555 patients with PHA, 22 A/KPA and only 2 cases of combined secretion were identified in unilateral nodular hyperplasia and adenomas of both adrenal glands [12]. The average diameter of the A/CPA is 26.2 mm, while the size of the aldosterome does not exceed 15 mm [11, 48]. These data are consistent with the fact that corticosteromes tend to be larger than aldosteromes, reaching an average of 3.5 cm in diameter [37, 49].
Thirdly, the explanation may simply be a more thorough assessment of the hormonal activity of formations when they are large (more than 2 cm) [14]. It is possible that the prevalence of subclinical hypercortisolism would be higher in smaller APAs if it were diagnosed more often.
Hybrid steroids in the diagnosis of primary hyperaldosteronism
Among the alternative laboratory differential diagnostic studies for APA and IHA, especially for tumors with combined secretion, the determination of con-steroids (CS) is relevant. Compounds such as 18-hydroxycortisol (18OHF) and 18-oxocortisol (18oxoF) have gained attention after they were shown to be increased in urinary excretion in PHA. The term “hybrid” reflects chemical transformations characteristic of both the glomerular (18-hydroxylation and 18-oxidation) and zona fasciculata (17-hydroxylation) adrenal cortex [41]. Both metabolites are detected in small quantities in healthy people [50] and their concentration increases significantly in familial forms of hyperaldosteronism (FA) types 1 and 3 [51, 52]. 18oxoF has gluco- and mineralocorticoid effects, many times less than aldosterone, while 18OHF is completely devoid of biological activity [53, 54]. The formation of hybrid steroids is catalyzed by enzymes of the final stages of the synthesis of gluco- and mineralocorticoids. 11β-hydroxylase (CYP11B1), located in the zona fasciculata, also has 18-hydroxylase activity. In patients with complete inactivation of aldosterone synthase, while maintaining high CYP11B1 activity, high concentrations of 18-hydroxycorticosterone are found, despite severe aldosterone deficiency [55]. Consequently, 11β-hydroxylase converts cortisol to 18-hydroxycortisol but not to 18-oxocortisol, whereas aldosterone synthase (CYP11B2), located exclusively in the zona glomerulosa, is able to carry out both conversions with greater efficiency [56–58]. Aldosterone synthase and 11β-hydroxylase are present in small quantities not only in the adrenal glands, but also in other tissues (smooth muscle, endothelium, brain), but their role in the synthesis of hybrid steroids seems to be very insignificant [59, 60]. Thus, cortisol can be converted to 18OHF by both aldosterone synthase in the zona glomerulosa and 11β-hydroxylase in the zona fasciculata. Since CYP17A expression is limited to the zona fasciculata, it is the main site of cortisol production. Therefore, the presence of 18OHF in healthy people is understandable. However, the synthesis of 18oxoF requires an additional step - 18-oxidation, which is possible only under the influence of aldosterone synthase, present exclusively in the zona glomerulosa, where cortisol synthesis does not occur. Since the blood supply to the adrenal gland is centripetal in nature (from the periphery to the center), and the passage of cortisol from the zona fasciculata to the glomerular gland is unlikely, the synthesis of 18-oxocortisol in the unchanged adrenal gland remains unexplained. E. Freel et al. [56] found a positive correlation between urinary excretion of hybrid steroids and the dose of hydrocortisone used in patients with decreased intrinsic cortisol secretion. Thus, the possibility of synthesizing 18OHF and 18oxoF from cortisol circulating in the blood was proven. One theory explains the possible effect of aldosterone synthase on cortisol by its movement in areas of “penetration” of the zona glomerulosa tissue into the fasciculata along the walls of small vessels. Be that as it may, a more complex synthesis pathway, requiring the indispensable participation of enzymes from different functional zones of the adrenal gland, explains the fact that in healthy people, the excretion of 18OHF is approximately 10 times higher than the excretion of 18oxoF [61].
High concentrations of hybrid steroids are typical for patients with familial PHA types 1 and 3 [51, 52]. Familial hyperaldosteronism type 1 (FHA-1), also known as glucocorticoid-suppressed, is a rare form of PHA inherited in an autosomal dominant pattern. The disease is caused by a mutation, as a result of which two genes ( CYP11B1
and
CYP11B2
), encoding 11β-hydroxylase and aldosterone synthase, respectively, a common chimera gene is formed [44].
The chimera gene contains a proximal region of CYP11B1
and a distal
region of CYP11B2
. The product of this gene is involved in the synthesis of aldosterone, but the activity of the gene is controlled by ACTH, and not by the renin-angiotensin system. As a result, aldosterone begins to be produced by the zona fasciculata, where only glucocorticoids are normally synthesized. Under the influence of a chimeric gene, the synthesis of hybrid steroids increases in the zona fasciculata [62]. Patients undergo lifelong therapy with glucocorticoids to suppress ACTH-dependent aldosterone production [63]. The diagnosis of GAS-1 can be confirmed by detecting the chimeric gene using PCR [64].
In 2008, a case of familial hyperaldosteronism type 3 (FHA-3) was first described, characterized by a significant increase in aldosterone concentration, severe hypokalemia, early onset of arterial hypertension, resistant to drug therapy, including potassium-sparing diuretics (spironolactone and amiloride) , which required bilateral adrenalectomy [52]. Other distinctive features of SGA-3 were pronounced bilateral adrenal hyperplasia, increased concentrations of hybrid steroids and, in response to the dexamethasone test, a paradoxical increase in plasma concentrations of aldosterone and hybrid steroids, no decrease in cortisol concentrations, and an increase in blood pressure. The disease is caused by a germline mutation of the KCNJ5
, located on chromosome 11q24 and encoding G protein-coupled inward rectifier potassium channel 4 (GIRK4 - from the English G protein-activated inward rectifier potassium channel 4, or Kir 3.4).
The mutation causes a loss of ion selectivity of the K+ channel and depolarization of the membrane, which leads to the opening of voltage-dependent Ca2+ channels, the influx of calcium ions into the cell, increased expression of CYP11B2
and increased aldosterone synthesis [65].
The concept of GS as biochemical markers of APA is based on the fact that their concentration in serum and urine in individuals with aldosteroma is higher than in patients with PHA and bilateral adrenal hyperplasia [66]. One theory explains the increased synthesis of GS in APA by their intra-adrenal location. Unlike the intact zona glomerulosa, the APA is not located on the outside of the adrenal gland, which allows it to receive blood from the surrounding zona fasciculata, which has a high concentration of cortisol, which is converted into 18oxoF by the tumor aldosterone synthase. On the other hand, as noted above, in the APA tissue there are cells that are phenotypically similar to cells of the zona fasciculata and are capable of coexpressing CYP11B1
,
CYP11B2
,
CYP17A1
, and therefore capable of producing cortisol [67, 68]. It is assumed that GS are formed in the tumor both from blood cortisol and locally in the tumor tissue. Thus, the loss of clear separation of functional zones in the adrenal tumor in patients with APA leads to the fact that cortisol can be affected by aldosterone synthase and, as a consequence, to increased synthesis of C18-oxidized steroids. The concentration of the latter depends on the concentration of cortisol in the blood and, more importantly, on the characteristics of the tumor cells.
High-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) can detect even very low concentrations of GS in peripheral plasma (lower limit for 18oxoF is 0.25 ng/dL) [69]. F. Satoh et al. [70] in 2015 showed for the first time that measuring the concentration of 18oxoF in peripheral blood using HPLC-MS/MS allows differentiating APA from IGA. Patients with confirmed PHA based on the results of SVBD and MSCT were preliminarily divided into patients with APA ( n
=113), IHA (
n
=121) and micro-APA (
n
=27) (in the latter subgroup, MSCT showed no changes in NP, but in CVBD, unilateral hyperproduction of aldosterone was determined) [70]. In all cases of APA and micro-APA, the diagnosis was confirmed after adrenalectomy based on the results of measuring plasma aldosterone concentrations and immunohistochemical analysis of steroid enzymes in the removed lesions [42, 71]. According to the results of the study, the concentration of 18oxoF in plasma turned out to be a more reliable differential diagnostic criterion for APA and IHA than the concentration of 18OHF. This can be explained by the presence of all the necessary components for the synthesis of the latter even in the tissue of the unchanged adrenal gland and, on the contrary, the need for enzymes from different zones for the production of 18oxoF [70]. In patients with APA, 18oxoF concentrations greater than 6.1 ng/dL and plasma aldosterone concentrations greater than 32.7 ng/dL were detected in 95 of 113 (84%) patients. Such indicators were not determined in any case of IHA. At the same time, in 43% of cases of IHA, the concentration of 18oxoF in plasma did not reach 1.2 ng/dL, the lowest value in patients with APA. In 30 of 121 patients with IHA, CT revealed masses in one of the adrenal glands, but they did not have hormonal activity. Therefore, measuring 18oxoF concentrations in such patients in settings where SIDS is not available may help avoid unnecessary surgery. The highest concentrations of 18oxoF and aldosterone in plasma in patients with micro-APA were 4.8 and 24.5 ng/dl, respectively, and were comparable to those in IHA, and therefore the authors emphasize the importance of CVBD for the differential diagnosis of these forms of the disease in patients with an aldosterone concentration <32.7 ng/dL and an “intermediate” concentration value of 18oxoF. Based on these results, it is proposed to include measurement of 18oxoF concentration in the diagnostic regimen for PHA [70]. However, it is noted that the concentration of 18OHF, while having less sensitivity, is characterized by high specificity comparable to 18oxoF and therefore can also be used in the differential diagnosis of APA and IHA [70]. Although measurements of plasma 18oxoF concentrations using HPLC-MS/MS are relatively expensive (approximately US$150 per sample), they are much less expensive than CVDS (approximately US$10,000) [70].
Confirmation of the increased synthesis of GS in the APA was the detection of a high concentration of 18oxoF in the blood of the adrenal veins. Y. Nakamura et al. [69] used the 18oxoF/cortisol ratio instead of aldosterone/cortisol during CVAD and showed that the concentration of the hybrid steroid and its ratio to cortisol is higher in blood flowing from the adrenal gland with APA than from the contralateral intact adrenal gland or from patients with IHA. Although the calculation of the 18oxoF/cortisol ratio has not been shown to be superior to the calculation of the aldosterone/cortisol ratio in the diagnosis of unilateral adrenal hyperfunction, the very fact of increased concentrations of 18oxoF in the blood flowing from the APA argues for the use of the GS determination in the diagnosis of PHA.
Thus, patients with APA have higher concentrations of GS, especially 18oxoF, than patients with bilateral adrenal hyperplasia. Although potential crossover between PHA subtypes limits the usefulness of the GS definition for the differential diagnosis of forms of PHA, such definitions can significantly narrow the indications for SIBD in patients planning surgical removal of the aldosteroma.
Features of aldosteres caused by somatic mutations
Somatic mutations underlying sporadic (non-familial) forms of PHA have been identified in at least 5 genes: KCNJ5
[65],
ATP1A1
,
ATP2B3
[72],
CACNA1D
[73], and
CTNNB1
[74].
According to a large European study, the prevalence of mutations in the first 4 genes among patients with APA was 54%, of which 38% were mutations in KCNJ5
[75].
A recent meta-analysis showed that the overall prevalence of KCNJ5
is 43% and is unevenly distributed between Europe, USA, Australia (up to 35%) and China, Japan (up to 63%) [76].
In contrast to the mutation in the KCNJ5
mutations in
ATP1A1
,
ATP2B3
and
CACNA1D
, which stimulate aldosterone production, were also found in clusters of aldosterone-producing cells of intact adrenal glands in kidney donors [77].
In a retrospective study in 2016, it was shown that some somatic mutations in APA can be determined with high accuracy (in 92% of cases) by a characteristic “7-steroid profile” (HPLC-MS/MS method): concentration of aldosterone, 21-deoxycortisol , corticosterone, 11-deoxycorticosterone, cortisol, as well as 18-oxocortisol and 18-hydroxycortisol [78]. APA with mutations in the ATPase or CACNA1D
is characterized by the cellular composition of the zona glomerulosa [45, 79].
APAs with mutations in KCNJ5
contain cells similar to zona fasciculata cells and are characterized by a higher concentration of GS in peripheral venous blood plasma.
Such APAs have a high level of expression of zona fasciculata enzymes – 11β-hydroxylase [45] and 17α-hydroxylase [80], which is manifested by increased production of cortisol compared to other APAs, and hence hybrid steroids synthesized from it using CYP11B2 [51]. As already noted, the share of APAs carrying mutations in KCNJ5
accounts for about 40%, and in East Asia the prevalence of such APAs reaches 73% [81]. Therefore, in this population, the determination of 18-oxocortisol is important for diagnosing the hyperaldosteronism subtype [70].
Thus, an increase in GS concentration is detected not only in familial forms of hyperaldosteronism and is due to the characteristics of steroidogenesis in APA cells. The heterogeneity of APA cells capable of co-expressing various enzymes of steroidogenesis indicates that an increase in the concentration of 18OHF and 18oxoF is a hallmark of sporadic APA, more often in the presence of certain somatic mutations, and the determination of GS can become an additional method for the non-invasive differential diagnosis of APA from IHA and hormonally inactive adenomas.
Hybrid steroids in the diagnosis of aldosterone- and cortisol-producing adenomas
When studying somatic mutations in A/KPA, KCNJ5
and
PRKACA
(protein kinase A catalytic subunit gene) [82, 83].
The latter is characteristic of corticosteras and is the most common mutation responsible for increased cortisol synthesis, the prevalence of which in the Chinese population is about 40% [84]. In 22 patients with A/KPA, 17 samples were identified containing the KCNJ5
and no cases of other mutations leading to hyperaldosteronism or hypercortisolism [12].
KCNJ5
mutations are common in both APA and cosecretory adenomas, while the prevalence of
PRKACA
was found in only 1.6% of cases among 122 patients with APA.
Therefore, A/CPA has more in common with aldosteroma than with corticosteroma [9, 83]. However, W. Arlt et al. [9] did not find a connection between the degree of cosecretion of glucocorticosteroids, the form of PHA and the presence of mutations in KCNJ5
, on the basis of which it can be assumed that hypersecretion of cortisol, due to the number of cells capable of its synthesis and their proportion in the total tissue mass, does not have a clear connection with one or another specific mutation.
Interestingly, in a study by H. Willenberg et al. CYP11B1/CYP11B2 was not detected
, and aldosterone synthase mutations and family history were excluded. Therefore, it is assumed that an increase in GS concentration is also typical for adenomas with combined secretion. It follows that with A/CPA, not only the results of CVID are distorted, but also the specificity of an increase in GS concentration as a sign of familial forms of PHA is reduced. This makes genetic PCR testing mandatory for diagnosis.
At the same time, confirmation of the possibility of using HS as biochemical markers of the combination of hyperaldosteronism and hypercortisolism contributes to the formation of a “picture” of the PHA subtype and the choice of appropriate treatment tactics without CVBD (see table).

Differential diagnosis of subtypes of primary hyperaldosteronism (PHA) with the determination of hybrid steroids (proposed algorithm based on analysis of literature data)