В этой статье мы расскажем вам о том, что такое неонатальный скрининг новорожденных, для чего его делают. Ведь многих мам волнует вопрос, зачем малышу в роддоме берут кровь из пяточки?
После рождения малыш вступает в жизнь с непривычными для него условиями, нуждаясь в нашей заботе и обереге от заболеваний. Частично свое чадо от «летающих» вирусов и бактерий мы защищаем. Однако существуют недуги, с которыми мы не в силах бороться при помощи витаминок или соблюдения режима дня.
Речь идет о наследственных или врожденных заболеваниях, не выявленных у малыша во время внутриутробного развития. Подробнее о генетических исследованиях до и во время беременности .
Для решения проблемы проводится неонатальный скрининг новорожденных — исследование новорожденного ребенка в первые дни жизни.

Что такое неонатальный скрининг
Неонатальный скрининг — массовое обследование всех новорожденных детей для раннего выявления наиболее распространенных наследственных или врожденных заболеваний.
Цель: обнаружение у малыша недуга до появления клинических признаков и назначение советующего своевременного лечения.
Правовое регулирование скринингового исследования новорожденных в Российской Федерации
Начиная с 2006 года, в рамках национального проекта «Здоровье» всем новорожденным детям проводят исследование на выявление врожденного гипотиреоза, фенилкетонурии, муковисцидоза, адреногенетального синдрома и галактозмии.
Ранее на протяжении 15 лет проводилось тестирование лишь на врожденный гипотиреоз и фенилкетонурию.
Порядок и сроки выполнения скрининга регламентируются Приказом Минздравсоцразвития России от 22.03.2006 г. № 185 «О массовом обследовании новорождённых детей на наследственные заболевания».
С 2007 года проводится аудиологический скрининг, рано выявляющий нарушения слуха у ребенка первого года жизни.
По Свердловской области перечень расширен до 16 патологий на основании Приказа МЗ СО от 02.03.2012 г. № 166-п «О совершенствовании массового обследования новорожденных детей на наследственные заболевания на территории Свердловской области».
Дополнительные одиннадцать заболеваний:
- Лейциноз
- Тирозинемия первого типа
- Цитруллинемия
- Множественная карбоксилазная недостаточность
- Недостаточность очень длинных цепей ацил-СоА-дегидрогеназы жирных кислот
- Недостаточность средних цепей ацил-СоА-дегидрогеназы жирных кислот
- Недостаточность митохондриального трифункционального белка/ недостаточность длинных цепей гидроксил-СоА-дегидрогеназы жирных кислот
- Глютаровая ацидурия первого типа
- Изовалериановая ацидемия
- Метилмалоновая ацидемия
- Пропионовая ацидемия
Определение заболевания
Фенилкетонурия ― это врожденная, генетическая патология, подразумевающая нарушения гидроксилирования фенилаланина. Характеризуется накоплением в организме аминокислоты и продуктов ее метаболизма, что ведет к тяжелым поражениям центральной нервной системы. Впервые заболевание было описано норвежским врачом И. А. Феллингом в 1934 году.
Изучая болезнь специалисты установили, что за наличие болезни отвечает единственный ген фенилаланингидроксилазы. Первое успешное лечение было разработано и проведено в 1950 году в Англии.

В неонатальном периоде клиника отсутствует. Патология проявляется в первые полгода жизни ребенка. В дальнейшем накопление вещества приводит к тяжелым нарушениям развития. Поэтому крайне важно сразу после рождения выявить дефект и не допустить употребление продуктов, содержащих фенилаланин. Более позднее соблюдение диеты не устранит полученные нарушения, но не допустит развития новых.
Патология одинаково часто встречается среди лиц обоих полов. Расовых особенностей не выявлено. Большое количество больных в таких странах как Китай, Турция, Ирландия. В среднем по России с фенилкетонурией рождается каждый 7-ми тысячный ребенок.
Сроки и условия проведения неонатального скрининга новорожденных
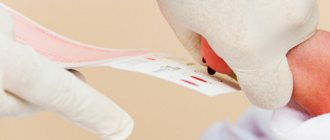
Исследование выполнятся в родильном доме на четвертый день жизни у доношенного ребенка, у недоношенного малыша — на седьмой или позднее. При тестировании на более ранних сроках возможно получение ложноположительных или ложноотрицательных результатов.
Набирается капелька крови из пятки малыша до кормления утром натощак либо чрез три часа после любого очередного кормления. Затем кровь наносится на тестовые бланки (фильтровальную бумагу) на область специальных кружочков, пропитанных специальным веществом.
Иногда скрининг не проводится в родильном доме — например, при выписке из стационара ранее четвертых суток жизни ребенка. Вам на руки выдается заполненный тест-бланк для забора крови, а в обменной карте указывается, что кровь на скрининг не взята. Набор крови проводится на участке согласно рекомендованным срокам.
Как расцениваются итоги исследования?
Результаты можно узнать в поликлинике по месту жительства через 10 дней после забора анализа.
Ребенок здоров при отрицательных результатах тестирования. Обычно родителей об этом не извещают. Наблюдение за ребенком проводится по возрасту согласно рекомендованным срокам в плановом порядке.
Положительный или сомнительный результат не всегда означает, что ребенок болен.
Скрининг — лишь расчет вероятного риска наличия недуга у крохи. Для уточнения диагноза проводится повторный забор крови. При вновь повышенных показателях ребенка с родителями направляют в медико-генетическую консультацию для тщательного и детального обследования.
Причины фенилкетонурии
Существует три типа генетического отклонения, первый считается классическим, поскольку диагностируется в более чем 90% случаев. Второй и третий ― более редкая форма патологии. Симптоматика схожа во всех типах, заболевание приводит к умственной отсталости. При классической форме фенилкетонурии избежать этого можно диетотерапией, но атипичные варианты, к сожалению, коррекции не подлежат.
- Классическая фенилкетонурия (I тип) ― это низкая выработка фенилаланингидроксилазы (ФАГ), что приводит к собиранию в естественных жидкостях организма фенилаланина и продуктов его расщепления. Патология вызвана мутированным геном РАН.
- Фенилкетонурия II типа ― недостаток дигидроптеридинредуктазы, что препятствует преобразованию фенилаланина в тирозин. Патология из-за мутации гена QDPR.
- Фенилкетонурия III типа ― недостаток 6-пирувоилтетрагидроптеринсинтазы, нужной для синтеза тетрагидробиоптерина. Патология вызвана мутированным PTS-геном.
Все формы заболевания наследуются по аутосомно-рецессивной форме. Это означает, что генетический дефект может быть унаследован у одного из родителей. Половая принадлежность родителя и ребенка не играет роли.
Классификация
Фенилкетонурия в настоящее время не имеет общепризнанной мировой классификации. Над этим вопросом ведутся дебаты, наравне с изучением заболевания. Чуть ранее, до расшифровки генов, считалось, что степень поражения интеллектуальных способностей зависит от степени активности фермента. Поэтому текущая квалификация признана устаревшей. Не учитывает она и другие симптоматические факторы.
При диагностировании ставят:
- I тип (дефицит ФАГ) ― концентрация ФА больше 20 мг/дл.
- Средняя форма ФКУ ― ФА от 8,1 до 20 мг/дл.
- Легкая форма ГФА-уровень ― ФА от 2,1 до 8,0 мг/дл.
При уровне до 8,0 мг/дл фенилкетонурию считают доброкачественной. Она не требует специального лечения, но необходимо наблюдение первый год жизни ребенка. Контролирует состояние врач-педиатр, невролог, генетик.
Выделяют также еще одну форму фенилкетонурии, не требующую коррекции. Это транзиторная форма ГФА в период новорожденности. Возникает, как правило, при недоношенности, что обусловлено функциональной незрелостью организма. Транзиторная фенилкетонурия ― это временное повышение ФА-уровня, способное подняться до критических значений. При этом клиника отсутствует либо проявления весьма незначительны. Через несколько месяцев биохимические показатели приходя в норму.
Патогенез
Механизм зарождения и развития фенилкетонурии связан с нарушением обмена органического соединения ― аминокислоты фенилаланина. Метаболический блок препятствует преобразованию фенилаланина в тирозин. Аминокислота не только не преобразуется, а накапливается в виде токсичных метаболитов:
- фенилмолочная кислота;
- фенилпировиноградная кислота;
- фенилуксусная кислота;
- фенилэтиламин и прочее.
Скопление фенил-веществ оказывает токсическое действие на ЦНС. В настоящий момент механизм еще до конца не изучен, врачам не известен патогенез дисфункции головного мозга.
Существуют предположения, что поражение нервной системы является результатом ряда факторов. Среди них как прямое токсического воздействие фенилаланина, так и нарушение обмена белков, липопротеидов и гликопротеидов, сбой гомонального метаболизмеа и мембранного транспорта аминокислот. Все это в комплексе имеет важное значение для созревания и правильного функционирования ЦНС.
Лечение
Симптоматическая терапия при любой формой фенилкетонурии неэффективна. Существует только один способ предотвратить негативные последствия заболевания ― диетотерапия. Из рациона исключают высокобелковые и содержащие фенилаланин продукты. Недостающее количество белка восполняют специализированным лечебным питанием, с максимально низким содержанием аминокислоты ФА или полностью ее лишенным. Следует учитывать, что эффективность терапии напрямую зависит от времени начала коррекции и уже произошедших патологических изменений.
Цель лечебного питания при классической форме заболевания ― это предотвращение развития нарушений ЦНС, физического и умственного развития. Легкая форма ГФА допускает расширение диеты под строгим наблюдением врача за состоянием ребенка и биохимическими показателями. Под запретом: мясо, рыба, орехи, шоколад и бобовые, все виды яиц, творог и сыры. Также следует исключить продукты, содержащие искусственный подсластитель аспартам.
Критерий эффективности лечения ― уровень ФА в крови.
Симптомы
I тип. Первые признаки у ребенка проявляются в возрасте от 2 месяцев до полугода.
- Апатичность либо, наоборот, повышенная раздражительность.
- Отсутствие интереса к окружению, людям, предметам, обстановке.
- Частое срыгивание.
- Аллергический дерматит.
- Нарушение мышечного тонуса.
- Пониженное давление.
- Судороги.
- Иногда развивается микроцефалия (малый размер черепа относительно других частей тела) и гидроцефалия (избыточная жидкость, омывающая головной мозг).
К характерным симптомам относятся гипопигментация кожи, волос, радужной оболочки глаз. Моча имеет специфический запах плесени или его еще называют «мышиным» запахом. Эпилептические припадки наблюдаются у половины больных, часто является первым выраженным клиническим симптомом. Приступ характеризуется «салаамовыми» судорогами, напоминающими кивки. Они случаются часто, плохо поддаются антиконвульсантному лечению.
Если не корректировать концентрацию ФА, болезнь прогрессирует. Как правило, уровень IQ у таких детей не превышает 20, при норме от 85. Умственная отсталость настолько сильная, что отсутствуют эмоциональные реакции, наблюдаются психопатии и шизофреноподобные расстройства.
II тип. Первая симптоматика проявляется на первом году жизни.
- Повышенная возбудимость.
- Задержка развития.
- Обильное слюнотечение.
- Сниженное артериальное давление.
- Частое повышение температуры тела.
- Сухожильная гиперрефлексия (повышение рефлексов) или спастический тетрапарез (обессиливание всех четырех конечностей).
- Миоклоническая эпилепсия (генерализованные приступы, преимущественно возникающие после пробуждения).
- Микроцефалия.
Отличительная особенность второго типа ― гибель нейронов, нарушение метаболизма фолатов, а также кальцификация в различных отделах головного мозга. Болезнь быстро прогрессирует, может привести к смерти ребенка в течение 2 — 3 лет.
III тип. Симптомы дефицита пирувоилтетрагидроптеринсинтетазы схож с проявлениями болезни Паркинсона:
- Постуральная нестабильность и трудности походки. Сложно либо невозможно поддерживать определенное положение всего тела или конечностей.
- Гипокинезия (низкая двигательная активность, ограниченный темп и объем движений).
- Гиперсаливация (повышенное слюноотделение).
- Нарушения глотания.
- Окулогирные кризы (симметричное отклонение обоих глаз, обычно направленное вверх).
В 80% случаев этот тип заболевания сопровождается снижением количества биогенных аминов в ликворе. Лечение затруднено тем, что раннее снижение концентрации ФА может вызвать серьезные патологические изменения. Несоблюдение диетотерапии приведет к замедлению развития речи, низкому интеллекту, проблемам с памятью.