In this article we will tell you about what neonatal screening of newborns is and why it is done. After all, many mothers are concerned with the question: why do they take blood from the baby’s heel in the maternity hospital?
After birth, the baby enters life with conditions that are unusual for him, needing our care and protection from diseases. We partially protect our child from “flying” viruses and bacteria. However, there are ailments that we cannot fight with the help of vitamins or adherence to a daily routine.
We are talking about hereditary or congenital diseases that were not detected in the baby during intrauterine development. Read more about genetic testing before and during pregnancy.
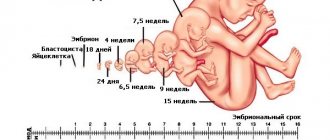
To solve the problem, neonatal screening of newborns is carried out - an examination of a newborn child in the first days of life.

What is neonatal screening
Neonatal screening is a mass examination of all newborn children for early detection of the most common hereditary or congenital diseases.
Goal: detecting an illness in a baby before the appearance of clinical signs and prescribing the appropriate timely treatment.
Legal regulation of newborn screening tests in the Russian Federation
Since 2006, as part of the national project “Health”, all newborn children have been tested to identify congenital hypothyroidism, phenylketonuria, cystic fibrosis, adrenogenetal syndrome and galactosmia .
Previously, for 15 years, testing was carried out only for congenital hypothyroidism and phenylketonuria.
The procedure and timing of screening are regulated by Order of the Ministry of Health and Social Development of Russia dated March 22, 2006 No. 185 “On mass screening of newborn children for hereditary diseases.”
Since 2007, audiological screening , early identifying hearing impairment in a child of the first year of life.
In the Sverdlovsk region, the list was expanded to 16 pathologies on the basis of Order of the Ministry of Health of the SD dated March 2, 2012 No. 166-p “On improving the mass examination of newborn children for hereditary diseases in the Sverdlovsk region.”
Additional eleven diseases:
- Leucinosis
- Tyrosinemia type 1
- Citrullinemia
- Multiple carboxylase deficiency
- Deficiency of very long chain fatty acid acyl-CoA dehydrogenase
- Medium chain fatty acid acyl-CoA dehydrogenase deficiency
- Mitochondrial trifunctional protein deficiency/long chain hydroxyl-CoA dehydrogenase fatty acid deficiency
- Glutaric aciduria type 1
- Isovaleric acidemia
- Methylmalonic acidemia
- Propionic acidemia
Definition of disease
Phenylketonuria is a congenital, genetic pathology that involves impaired hydroxylation of phenylalanine. It is characterized by the accumulation of amino acids and the products of its metabolism in the body, which leads to severe damage to the central nervous system. The disease was first described by the Norwegian doctor I. A. Felling in 1934.
While studying the disease, experts found that a single phenylalanine hydroxylase gene is responsible for the presence of the disease. The first successful treatment was developed and carried out in 1950 in England.

There is no clinic in the neonatal period. The pathology manifests itself in the first six months of a child’s life. Subsequent accumulation of the substance leads to severe developmental disorders. Therefore, it is extremely important to identify the defect immediately after birth and prevent the consumption of foods containing phenylalanine. Later adherence to the diet will not eliminate the resulting disorders, but will not allow the development of new ones.
The pathology is equally common among both sexes. No racial characteristics were identified. A large number of patients are in countries such as China, Türkiye, and Ireland. On average, in Russia every 7 thousandth child is born with phenylketonuria.
Terms and conditions for neonatal screening of newborns
The study will be performed in the maternity hospital on the fourth day of life for a full-term baby, and for a premature baby - on the seventh or later. When testing at earlier stages, it is possible to obtain false positive or false negative results.
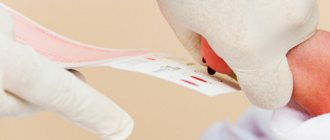
A drop of blood is collected from the baby’s heel before feeding in the morning on an empty stomach or three hours after any regular feeding. Then the blood is applied to test forms (filter paper) in the area of special circles soaked in a special substance.
Sometimes screening is not carried out in the maternity hospital - for example, when discharged from the hospital before the fourth day of the child’s life. You will be given a completed test form for blood collection, and the exchange card will indicate that no blood was taken for screening. Blood collection is carried out at the site according to the recommended time frame.
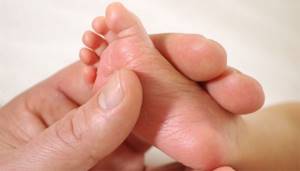
How are the results of the study assessed?
The results can be found at your local clinic 10 days after the test is taken.
The child is healthy with negative test results. Usually parents are not notified about this. Observation of the child is carried out according to age according to the recommended periods in a planned manner.
A positive or questionable result does not always mean that the child is sick.
Screening is just a calculation of the likely risk of a baby having a disease. To clarify the diagnosis, a repeat blood sample is taken. If the indicators are again elevated, the child and his parents are referred to a medical genetic consultation for a thorough and detailed examination.
Causes of phenylketonuria
There are three types of genetic disorder, the first is considered classic, since it is diagnosed in more than 90% of cases. The second and third are a rarer form of pathology. Symptoms are similar in all types, the disease leads to mental retardation. In the classic form of phenylketonuria, this can be avoided by diet therapy, but atypical variants, unfortunately, cannot be corrected.
- Classic phenylketonuria (type I) is a low production of phenylalanine hydroxylase (PAH), which leads to the accumulation of phenylalanine and its breakdown products in natural body fluids. The pathology is caused by a mutated RAS gene.
- Phenylketonuria type II is a deficiency of dihydropteridine reductase, which prevents the conversion of phenylalanine to tyrosine. Pathology due to mutation of the QDPR gene.
- Phenylketonuria type III is a deficiency of 6-pyruvoyltetrahydropterin synthase, necessary for the synthesis of tetrahydrobiopterin. The pathology is caused by a mutated PTS gene.
All forms of the disease are inherited in an autosomal recessive manner. This means that the genetic defect can be inherited from one of the parents. The gender of the parent and child does not play a role.
Classification
Phenylketonuria currently does not have a generally accepted worldwide classification. There is ongoing debate over this issue, as well as research into the disease. A little earlier, before deciphering genes, it was believed that the degree of damage to intellectual abilities depended on the degree of enzyme activity. Therefore, the current qualification is considered outdated. It also does not take into account other symptomatic factors.
When diagnosing they put:
- Type I (PAH deficiency) - FA concentration is more than 20 mg/dl.
- The average form of PKU is FA from 8.1 to 20 mg/dl.
- Mild form of HPA level - FA from 2.1 to 8.0 mg/dl.
At levels up to 8.0 mg/dL, phenylketonuria is considered benign. It does not require special treatment, but observation is necessary for the first year of the child’s life. The condition is monitored by a pediatrician, neurologist, and geneticist.
There is also another form of phenylketonuria that does not require correction. This is a transient form of HPA during the neonatal period. Occurs, as a rule, with prematurity, which is due to the functional immaturity of the body. Transient phenylketonuria is a temporary increase in FA levels that can rise to critical values. In this case, there is no clinical manifestations or the manifestations are very minor. After a few months, biochemical parameters returned to normal.
Pathogenesis
The mechanism of the origin and development of phenylketonuria is associated with a violation of the metabolism of an organic compound - the amino acid phenylalanine. The metabolic block prevents the conversion of phenylalanine to tyrosine. The amino acid not only is not converted, but accumulates in the form of toxic metabolites:
- phenyllactic acid;
- phenylpyruvic acid;
- phenylacetic acid;
- phenylethylamine and so on.
The accumulation of phenyl substances has a toxic effect on the central nervous system. At the moment, the mechanism has not yet been fully studied; doctors do not know the pathogenesis of brain dysfunction.
There are suggestions that damage to the nervous system is the result of a number of factors. Among them are both the direct toxic effects of phenylalanine and disruption of the metabolism of proteins, lipoproteins and glycoproteins, failure of homonal metabolism and membrane transport of amino acids. All this together is important for the maturation and proper functioning of the central nervous system.
Treatment
Symptomatic therapy for any form of phenylketonuria is ineffective. There is only one way to prevent the negative consequences of the disease - diet therapy. High-protein foods and phenylalanine-containing foods are excluded from the diet. The missing amount of protein is replenished with specialized therapeutic nutrition, with the lowest possible content of the amino acid FA or completely devoid of it. It should be taken into account that the effectiveness of therapy directly depends on the time the correction began and the pathological changes that have already occurred.
The goal of therapeutic nutrition in the classic form of the disease is to prevent the development of disorders of the central nervous system, physical and mental development. A mild form of HFA allows for an expansion of the diet under the strict supervision of a doctor over the child’s condition and biochemical parameters. Prohibited: meat, fish, nuts, chocolate and legumes, all types of eggs, cottage cheese and cheeses. You should also avoid foods that contain the artificial sweetener aspartame.
The criterion for the effectiveness of treatment is the level of FA in the blood.
Symptoms
Type I The first signs in a child appear between the ages of 2 months and six months.
- Apathy or, conversely, increased irritability.
- Lack of interest in the environment, people, objects, settings.
- Frequent regurgitation.
- Allergic dermatitis.
- Violation of muscle tone.
- Low pressure.
- Cramps.
- Sometimes microcephaly (small size of the skull relative to other parts of the body) and hydrocephalus (excess fluid surrounding the brain) develop.
Characteristic symptoms include hypopigmentation of the skin, hair, and iris. Urine has a specific musty smell or is also called a “mouse” smell. Epileptic seizures are observed in half of the patients and are often the first pronounced clinical symptom. The attack is characterized by “salaam” convulsions, reminiscent of nodding. They occur frequently and do not respond well to anticonvulsant treatment.
If PA concentrations are not adjusted, the disease progresses. As a rule, the IQ level in such children does not exceed 20, with the norm being 85. Mental retardation is so severe that there are no emotional reactions, psychopathy and schizophrenia-like disorders are observed.
Type II The first symptoms appear in the first year of life.
- Increased excitability.
- Developmental delay.
- Profuse drooling.
- Reduced blood pressure.
- Frequent increase in body temperature.
- Tendon hyperreflexia (increased reflexes) or spastic tetraparesis (weakness of all four limbs).
- Myoclonic epilepsy (generalized seizures, predominantly occurring upon awakening).
- Microcephaly.
A distinctive feature of the second type is the death of neurons, disruption of folate metabolism, as well as calcification in various parts of the brain. The disease progresses rapidly and can lead to the death of a child within 2 to 3 years.
III type. Symptoms of pyruvoyltetrahydropterin synthetase deficiency are similar to those of Parkinson's disease:
- Postural instability and gait difficulties. It is difficult or impossible to maintain a certain position of the whole body or limbs.
- Hypokinesia (low motor activity, limited tempo and range of movements).
- Hypersalivation (increased salivation).
- Swallowing disorders.
- Oculogyric crisis (symmetrical deviation of both eyes, usually directed upward).
In 80% of cases, this type of disease is accompanied by a decrease in the amount of biogenic amines in the cerebrospinal fluid. Treatment is complicated by the fact that an early decrease in FA concentration can cause serious pathological changes. Failure to comply with diet therapy will lead to slower speech development, low intelligence, and memory problems.